Article: Dr. Helmut Priewer and Christoph Bloch
Oximes as Nerve Agent Antidotes
From Hydroxylamine to HI 6-DMS
Organophosphate-type nerve agents (e. g. tabun, sarin, soman, cyclosarin, VX) are among the most toxic synthetic compounds known so far. Organophosphate intoxication causes an excess of the neurotransmitter acetylcholine by inhibition of the acetylcholinesterase that normally reduce the activity of acetylcholine. The following study gives an insight in the nerve agent antidotes belonging to a family of compounds called oximes (pralidoxime, obidoxime, HI 6-DMS). They are reactivators of the acetylcholinesterase. Additionally, the study provides prospects for further development of autoinjectors containing nerve agent antidotes.
1. Nerve Agents
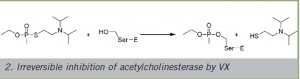 Irreversible inhibition of acetylcholinesterase by VX
Irreversible inhibition of acetylcholinesterase by VX
Organophosphate-type (OP) nerve agents are among the most toxic synthetic compounds known so far. They are chemically related to organophosphorous insecticides which frequently cause intoxications in civilian use. The work of the German chemist G. Schrader was the starting point of the development of nerve agents. In his search for plant protection agents in the second quarter of the 20th century he discovered the three highly potent compounds of tabun, sarin and soman which are also referred to as G-type agents in Germany due to their origin. Apart from these compounds especially cyclosarin and VX are of military significance.
Acetylcholine (ACh) is a neurotransmitter of agitational states in certain nerve cells. Signal transmission must be limited to avoid overstimulation. This is achieved by splitting the released ACh into acetic acid and choline by means of the acetylcholinesterase enzyme (AChE) to inhibit the stimulation of receptors. OP intoxications cause an excess of ACh since it is no longer degraded. Death is caused mainly by respiratory paralysis.
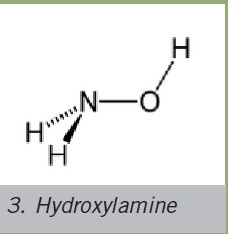 Hydroxylamine
Hydroxylamine
The AChE active center - of the serine hydrolase family - consists of a so called "anionic" binding site and a catalytic triad. While the quaternary, positively charged nitrogen of the ACh probably docks via cation-p-interactions into the "anionic" binding site - also referred to as choline pocket - thus sterically orienting the ACh molecule, the electrophile carbon atom of the acetyl remainder reacts biologically reversibly with the nucleophile hydroxyl group of the catalytic triad serine, resulting in the splitting of the ACh. In a further step the covalently bound acetyl remainder is hydrolyzed from the serine, rendering the enzyme functional again.
The "Tammelin ester" VX, a particularly toxic nerve agent with a structure similar to ACh also docks into the "anionic" binding site of the AChE with the nitrogen which is also positively charged after protonation. The structures of the tabun, sarin, soman and cyclosarin nerve agents are not similar to ACh. The joint effect of all nerve agents is the biologically irreversible reaction of the electrophile phosphorous atom of the OP with the hydroxyl group of the catalytic triad serine and thus the inhibition of the enzyme.
2. Oximes
The AChE-reactivating characteristics of the hydroxylamine acting as a nucleophile were proven in the early 1950s. However, this compound is not suitable for therapeutic purposes due to its high inherent toxicity.
Alternatively, aldoximes can be used as condensation products from hydroxylamine and aldehydes.
In addition to the C-N double bond aldoximes contain a hydrogen atom, an organic remainder, a hydroxyl group and a free electron pair of the nitrogen atom.
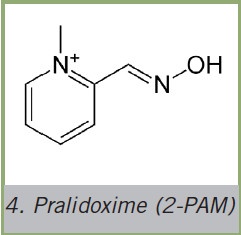 Pralidoxime (2-PAM)
Pralidoxime (2-PAM)
The reactivation of inhibited AChE brought about by nucleophile aldoximes, which will be referred to as oximes in the following, was optimized even further by means of a structure also similar to ACh - meaning the positively charged, quaternary nitrogen. Thus, the pralidoxime (2-PAM) - a quaternary monoxime - was developed mid-1950, providing positively charged nitrogen via a pyridinium aromatic ring. Quaternary pyridine oximes represent lead structures for acetylcholinesterase reactivators.
The effects of oximes are based on the chemical re-activation of the OP-inhibited AChE. The oximes dock into the choline pocket of the AChE active center with their quaternary, positively charged nitrogen, by their nucleophilic action attacking the inhibited enzyme at the phosphorous atom of the OP remainder via the oxygen atom of the oxime group. This results in an OP-oxime-compound whose rapid decomposition into secondary products is crucial for the effectivity of the reactivators. Since the oxime takes up the OP remainder the AChE is released and thus also regenerated.
In this context the "aging" of inhibited AChE is of particular significance since it nullifies the effects of oximes. For if another organic remainder (alcohol group) of the OP component of the inhibited enzyme is hydrolyzed the P-atom in turn is more difficult to attack by nucleophilic action which results in an undesired, highly stabilized OP-serine bond. The enzyme can then no longer be re-activated by oximes as it is an aged enzyme.
In the late 1950s Lüttringhaus and Hagedorn began studies on the synthesis of other oximes for the Bundeswehr at Freiburg University.
The LüH 6 oxime (obidoxime) was a therapeutic improvement. This diquarternary dipyridine oxime consists of two pyridine-4-aldoximes connected by a three-link dimethylether bridge (linker). The molecule duplication apparently resulted in increased effectiveness.
3. HI 6
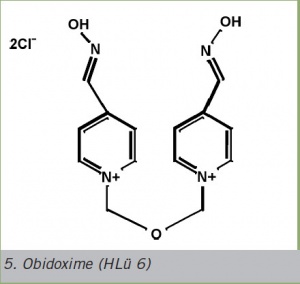 Obidoxime (HLü 6)
Obidoxime (HLü 6)
A further improvement of the re-activation of acetylcholinesterase in case of soman poisoning was achieved in the mid-1960s by means of the HI 6 oxime which was synthesized by Hagedorn.
This also involves a diquarternary dipyridine monoxime with a three-link dimethylether bridge which in contrast connects a pyridine-2-aldoxime (e. g. 2-PAM) with a pyridine-4-carboxamide.
While the pyridine-2-aldoxime ring is responsible for AChE re-activation at the OP-inhibited serine of the catalytic triad, the pyridine-4-carboxamide ring could cause the HI 6 to enter the choline pocket of the "anionic" bond site of the active center by means of an ACh-bio-isostere function. The required flexibility of the molecule would be ensured by the three-atom linker between the two quaternary ring systems.
HI 6 has the typical structure of the highly effective Hagedorn oximes, also referred to as H-oximes.
4. HI 6-DMS
In aqueous solutions HI 6 quickly decomposes due to its inherent catalytic ability. However, this also means that the military use of HI 6 is difficult since the oxime can be dissolved in water only shortly before its application. To this end, binary auto-injectors were developed, containing undissolved oximes and atropine solutions in separate chambers to be combined only shortly before an injection.
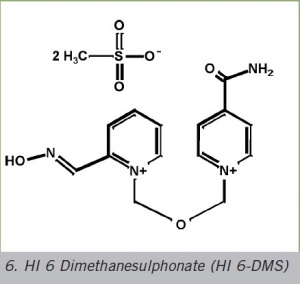 HI 6 Dimethanesulphonate (HI 6-DMS)
HI 6 Dimethanesulphonate (HI 6-DMS)
The problem with the HI 6 dichloride salt is that its solubility as well as its dissolution rate are too low to dissolve the quantity required for therapeutical purposes. This problem is solved by substituting the dimethanesulphonate (DMS) counter ion for the chloride. The solubility as well as the dissolution rate of the HI 6 - DMS are significantly improved.
This salt can be presented - also on an industrial scale - by means of a synthesis alteration by using bis(methanesulphonoxymethyl) ether instead of the extremely carcinogenic and genotoxic bis(chloromethyl)ether (BCME).
The Bundeswehr Medical Service intends to significantly improve treatment of nerve agent poisoning by developing and qualifying a combination auto-injector containing atropine sulphate, HI 6 DMS and a water-soluble benzodiazepine, e. g. Avizafon or Midazolam, for self- and buddy aid; HI 6 - DMS is intended to be developed and qualified for infusion applications for inpatient treatment.
After half a century of extensive military medical research and development for the Bundeswehr HI 6 - DMS is a relatively new development in the field of oximes, representing a further step towards an improved therapy of nerve agent poisoning.
Authors:
Pharmacy Director
Dipl.-Ing. Dr. phil. nat. Helmut Priewer
Pharmacist for Information on Drugs
Date: 05/09/2019
Source: Medical Corps International Forum (2/2011)


